
开篇语
假设检验,相信大伙儿或多或少都接触过,比如你的实验有两组(实验组、对照组)数据,你要分析一下它们之间的差异,那么假设检验是怎么也绕不过去的步骤。反正你不想做,你导师也会要求你做的,你导师不要求,你投稿时,那些reviewers也会要求你做的……
既然躲不开绕不去,那我们也不用怕它,小编今天就来跟大伙儿好好理一理,假设检验到底是啥东东,应该怎么玩!
基本概念与套路
什么是假设检验呢?从字面上看,肯定得先有个假设对吧,然后我们就来检验这个假设是不是成立,听上去很简单嘛!说得专业一点其实也不难理解,就是根据得到的数据,对我们预先设定的假设进行统计学检验,最后判断结果是否有统计学意义。
基本套路一般是:
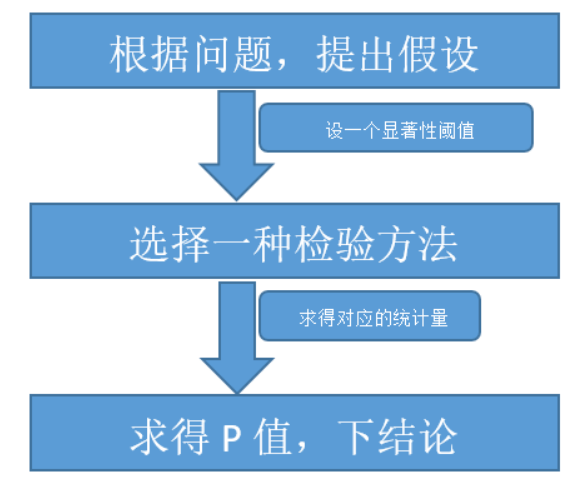
套路清楚后,我们现在来拆解套路:
根据问题 提出假设
什么是“假设(hypothesis)”呢?牛津词典中有这样的解释:A supposition or proposed explanation made on the basis of limited evidence as a starting point for further investigation。看着有点晕是不是?我们来个通俗的理解就是,当我们没有足够的证据(即先验知识)来说明一个问题时,我们可以先给出一个假定的说明。这个预先给出的假定说明,我们通常称为原假设或者零假设,与之对应的,我们还可以提出一个对立假设,又称为备择假设。
举个例子。当我们拿到一个硬币,想研究一下它的正反面是否均匀,我们可以先假设它是不均匀的,这个就是零假设;那么对应的,它正反面是均匀的,就是备择假设。
PS:一般来说(并不绝对),原假设就是需要我们收集证据来反对的假设,而备择假设就是需要收集证据来支持的假设。
扩展阅读 为啥有了零假设,我们还需要弄一个对应的备择假设呢?对备择假设的重要性,
90多年前,英国著名的统计学家哥色特(其笔名就是Student)曾举例解释过这个问题,
他的主要思想就是人们往往都倾向于选择相信概率比较大的事件。
比如一些来自于正态总体的数据,现想检验它们的均值是不是等于a0?
假设得到检验的概率值为0.0001,虽然这个值很小,但是你不能认为这批数据的均值不等于a0,为什么呢?
因为这时候你只有一个a0供你检验,概率值再小,也不能否认它发生的可能性。
而此时,如果你再有一个“备胎”(值为a1)让你去检验,最后检验的概率值为0.05,比前面的值大很多,
这时候你就会倾向于选择后面a1这个值,而认为原来的a0不真。
所以,我们需要有“比较”,多一个“备胎”,多一份选择!(此例子原型来源于《数理统计学简史》)。
“假设”通常有三种形式,以我们最熟悉的生物样本数据的差异分析来举例,当我们分析的是单个样本时,我们可以比较这个样本的数据与一个已知值的关系,是大于,小于,还是等于。
扩展阅读
单个样本的假设的形式如下所示(其中H0为原假设,H1为备择假设,θ0为已知的值):
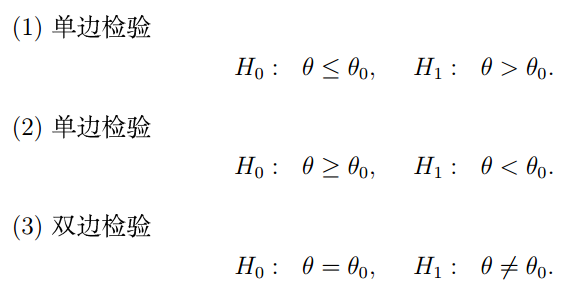
所谓单边检验,就是检验我们拿到的数据与一个已知值相比,是否大于等于它(或者小于等于它,你可以理解为,只需要判断数轴一侧的大小关系,所以叫单边检验),双边检验就是看两个值是否相等(你可以理解为,既不能大于,也不能小于,两边不等的情况都得排除,所以叫双边检验)。
当我们分析的是两个样本,考察它们之间是否存在差异,也有三种情况,就是样本1的值大于等于样本2,或者小于等于样本2,或者样本1与样本2相等。
扩展阅读
两个样本的假设的形式如下图(其中H0为原假设,H1为备择假设):

再有一种情况就是分析多个样本之间的差异,这时候就无所谓大于小于这些单边检验了,只有全部相等(或不全相等)的情况。
扩展阅读
多个样本之间的情况如下图所示:
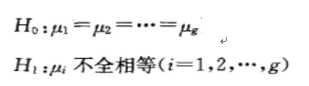
如果上面的解释和公式你还是觉得有点晕,那么我们来举个例子,以蛋白质定量数据为例,分析两组数据之间是否存在差异,我们假设现在有两大组数据(R, C),每一组得到5次生物学重复数据。得到数据如下:
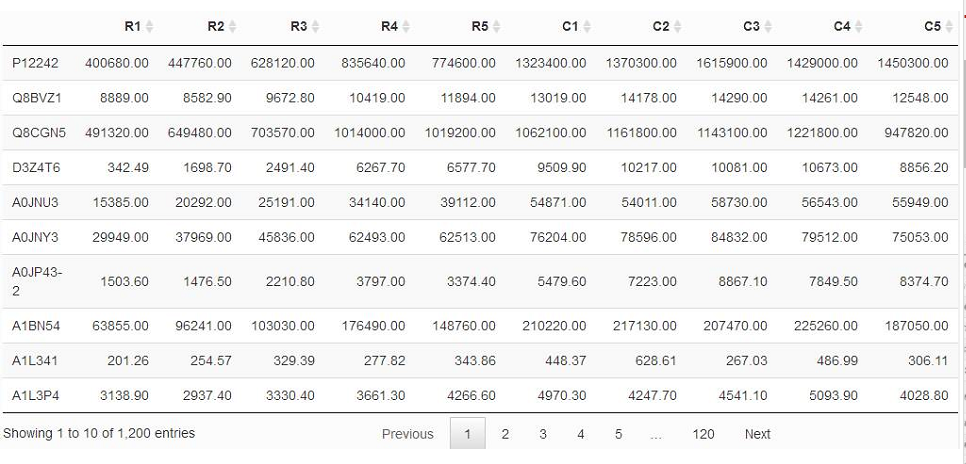
比如我们想看蛋白质P12242在R组和C组的平均表达量(分别设为μ1,μ2)是否一致,也就是说,要么相等,要么不等,大伙儿可以对照着看看上面提到的两个样本双边检验的说明。此时我们做原假设H0,即该蛋白质在两组中表达量相等(μ1=μ2),那么备择假设就是H1,即该蛋白质在两组中表达量不相等(有差异,即μ1≠μ2)。
假设给出来以后,我们一般会先选定一个显著性水平a,也就是一个判断的阈值,当统计分析出来的p值小于这个阈值时,说明是有统计显著性的,可以推翻原假设。通常可以设为0.05,严格一点可以设置为0.01,具体设多少要根据你的实验设计和目的,可以做一定的调整。
扩展阅读
为啥我们要设一个显著性水平a作为阈值呢?这就得说到假设检验中通常爱犯的两类错误(如下表):第一类错误(又称I型错误)是,否定了真实的原假设,即“弃真”,对应上面的例子就是,原假设是蛋白质P12242在两组中的表达量平均值是没有差异的,本来它是成立的,结果被你否定了,你认为有差异,这就产生了错误;第二类错误(又称II型错误)是,接受了错误的原假设,即“取伪”。怎么控制第一类犯错误的概率呢?就是我们通常说的设定一个显著性水平α,第二类犯错误的概率通常标记为β。

可以告诉你的是,在给定样本的情况下,减少犯其中一类错误的概率,就会导致增加犯另一类错误的概率。所以若想同时减少犯两类错误概率,就需要增加样本的容量。这就是为什么我们在分析差异表达的时候,通常都对样本量有一定的要求,否则你就算是做了这些假设检验,结果也比较难有说服力。当然,增加样本容量就会增加实验成本,所以通常建议在能力容许的范围内,尽可能的多增加一些样本量。
选择一种检验方法
好,我们继续说“蛋白质P12242在两组中的表达量平均值是没有差异”这个故事。上面的部分我们已经把假设列好了,原假设就是它们没差异,备择假设是它们有差异。接下来,我们就得开始检验这个假设了。
检验假设当然是需要有检验方法的!检验方法当然是不止一种的!针对这个例子,我们列出几种常用的假设检验的方法。做假设检验的一般思路就是,根据某种分布,求出数据对应的统计量,以此来判断该值是否落入拒绝域中(即拒绝原假设的取值范围),从而也可以得到对应的概率值P(以前可通过查表得到,现在通过计算机可以快速计算)。所以,接下来我们就以各种分布为主线,跟大伙儿聊聊假设检验的常见思路,详细的方法介绍我们会在下一篇推文中进行全面的总结。
正态分布和t分布(T检验)
正态分布,大伙儿从小就听说过吧!它也许是最常见,最重要的一种分布形式了。它虽不是著名数学家Gauss第一个提出的,但是他将之应用于天文学研究,使其广为人知,所以正态分布通常又称为高斯分布。
扩展阅读
正态分布的概率密度函数表达式为:
![]()
其中m和s为两个常数,m表征数据的均值,s表征数据的标准差。此时可称随机变量X服从参数为m, s的正态分布,记作X~N(m, s2)。

(其中红线表示的是均值为-2,方差为1的正态分布曲线,蓝线表示的是均值为2,方差为4的正态分布曲线。
从中可以看出,方差越小,图像越“瘦高”,方差越大,图形越“矮胖”。)
t分布是由英国著名统计学家哥色特发表,其笔名是“Student”,所以该分布又称为“Student t分布”。该分布的公布,标志着小样本统计推断的开始。
扩展阅读
其随机变量T的表达式为:

其中X~N(0, 1),Y~C-2-(n),且X,Y相互独立,其自由度为n,记作T~t(n)。
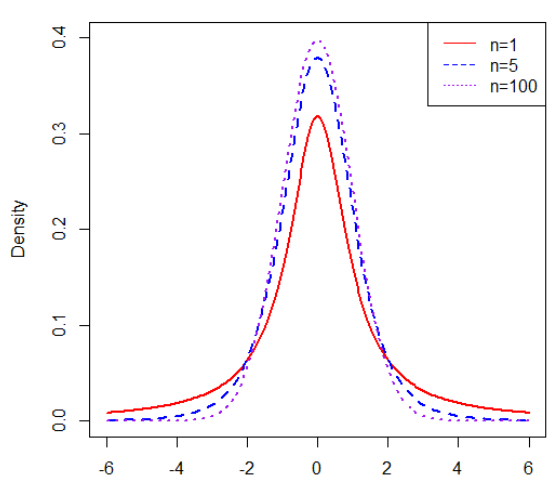
(红线、蓝线、紫线分别是自由度为1、5、100的T分布曲线。大家也许发现了,T分布与正态分布曲线看上去是不是很像,这里主要是因为T分布是为了小样本的数据分布,当自由度n趋于无穷大时,T分布曲线就越接近标准正态分布曲线。)
下面我们继续使用上面蛋白质定量数据的例子,来说明一下t.test函数的使用:
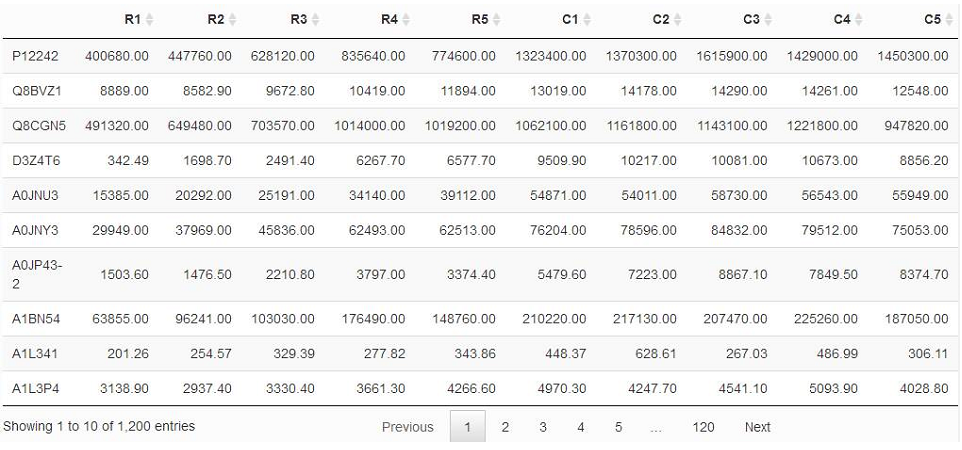
现在我们想检验蛋白质P12242在R组和C组的平均表达量(分别设为μ1,μ2)是否一致。我们已经设定好了原假设H0,即该蛋白质在两组中表达量相等(μ1=μ2),那么备择假设就是H1,即该蛋白质在两组中表达量不相等(有差异,即μ1≠μ2)。
然后我们选择一种检验方法,这里我们假设两组数据满足正态分布,所以我们选择用T检验来评估它们是否有差异(如何判断数据是否满足某种分布,这个问题后面我们会再讲)。
读入数据,
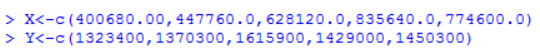
在R语言中可以直接使用t.test函数可进行T检验,函数原型为:

其中x, y是我们样本数据构成的向量,alternative表示备择假设的形式,“two.sided”表示双边检验(H1: μ1≠μ2), “less”表示单边检验(H1:μ1<μ2),“greater”表示单边检验(H1:μ1>μ2),paired表示样本是否是配对的,var.equal表示两总体方差是否相等。
在这个例子中,t.test函数调用如下:

最后得到P值等于0.0001212,小于0.05,可以支持我们拒绝原假设,即我们认为蛋白质P12242在R组和C组的平均表达量不相等。
通过以上的假设检验,我们就可以挑选出表达差异的基因/蛋白质,进行后续生物学意义上的注释、分类、聚类、相互作用关系分析等。
扩展阅读
怎么判断两样本是否配对?一般出现以下情况中的任意一种,就认为是配对的:
-
配对的两个受试对象分别接受两种不同的处理;
-
同一受试对象接受两种不同的处理;
-
同一受试对象处理前后的结果进行比较(即自身配对);
-
同一对象的两个部位给予不同的处理。
F分布(方差分析)
另一种常用来评估各数据组均数之间差异的方法叫方差分析,用这种方法,可以评估所有观测数据之间的波动程度,不同组之间的波动,以及组内数据的波动。
与方差分析相关的是F分布,由1924年英国统计学家R.A.Fisher提出,所以用Fisher的第一个字母F来命名。假设有两个独立的随机变量,这两个变量都分别符合卡方分布,它们相除以后的比率,我们就用F分布来描述。
扩展阅读
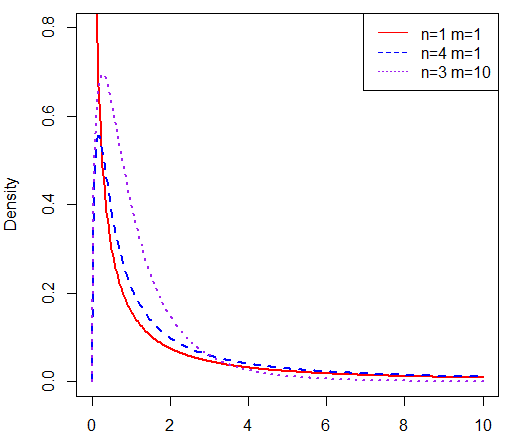
假设样本X~c2(n),Y~c2(m),且X和Y相互独立,则称随机变量F:
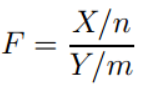
服从自由度为(n, m)的F分布,记作F~F(n, m)。使用R语言画出F分布曲线及R代码如下:

在R语言中,我们通常使用aov函数进行计算分析:

其中formula是方差分析的公式,data就是我们的数据集,其是数据框的形式。
formula中各符号表达的含义可以参考《R语言实战》一书,在使用的过程中要注意不要搞混了:

我们来举个例子说说aov的使用,比如分析小白鼠接种了3中不同菌型的伤寒杆菌后,平均存活的天数有无显著差异?原始数据如下:

代码实现:

从结果中得p值为0.0012,小于0.05,即认为3组小鼠的平均存活天数有显著性差异。
扩展阅读
在进行aov函数分析之前,我们一般要对数据进行正态性检验和方差齐性检验,在R语言中常用到的函数分别为shapiro.test和bartlett.test。只有满足要求的数据,我们才能进行该方法的计算。对于不满足要求的数据,我们可以采用秩和检验的方法,比如KW秩和检验(使用kruskal.test函数)或者Friedman秩和检验(使用friedman.test函数)。它们的详细使用我们计划在下一篇推文中详细介绍。
卡方分布(卡方检验)
上面我们讲了针对数据差异分析的检验方法,接下来跟大伙儿聊聊如何检验数据是否符合某种分布,包括对列联表的小样本量数据的检验。
话说,如果有n个相互独立的随机变量,它们都服从标准正态分布,那么这n个随机变量的平方和可以构成一个新的随机变量,这个新的随机变量分布的规律称为卡方分布。是不是很厉害的样子?
扩展阅读
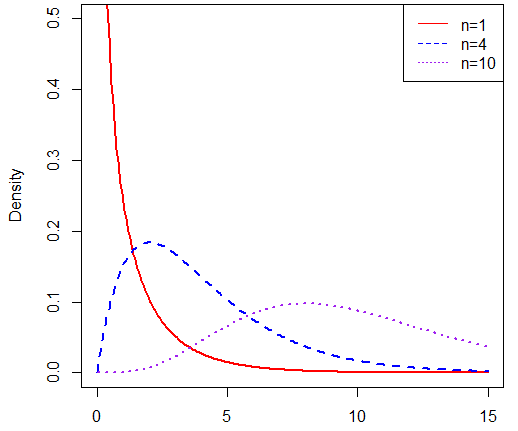
卡方分布的形式是这样的,我们假设样本Xi~N(0,1),其中i=1,2,3…n,则随机变量Y:
![]()
则称Y~c2(n),即Y服从自由度为n的卡方分布
R语言代码如下:

图形如下(其中红线、蓝线、紫线分别是自由度为1、4、10的卡方分布曲线),
如果以卡方分布为理论依据进行假设检验,这种检验方法我们称为卡方检验。
卡方检验主要用在两个方面:
a. 检验数据是否服从某种分布,比如卡方分布、均匀分布、泊松分布、正态分布等
b. 检验列联表数据。
Tips
啥叫列联表呢?其实就是交互分类表。交互分类是同时依据两个或多个变量的值,
将所研究的个案分类。交互分类的目的是将两个或者多个变量分组,
然后比较各组的分布状况,以寻找变量间的关系。比如下面这个列联表,
就是依据是否吸烟与是否患肺癌进行分类,然后可以通过比较这些数值,寻找吸烟与患肺癌之间是否相关。
R语言中使用chisq.test函数进行计算:

其中x, y为数据向量,correct表示是否进行连续性修正;p表示原假设落在某区间的理论概率,默认的是均匀分布;rescale.p=FASLE的意思是要求输入的p之和要等于1;simulate.p.value表示是否使用仿真的方法计算p值,B是指仿真的次数。
检验数据是否服从某种分布
说了这么多,到底我怎么检验我们的数据是符合什么分布呢?接下来我们就来举个例子。
我们都知道,不管是中考、高考、还是某类大型考试,某部门一般都爱要求老师给学生打的成绩要服从正态分布,那么现在假设我们得到了这样一批学生的成绩,我们想来检验下这批学生的成绩是否服从正态分布呢?
代码如下:

从结果中可得p值等于0.06161,大于0.05,可认为此次老师批阅试卷的成绩服从正态分布。
在检验数据服从某种分布时,我们还可以使用Kolmogorov-Smirnov检验,也是可以用于检验各种常见的分布,其函数是ks.test。比如还是上面那个例子,其代码实现如下:

得到的p值为0.2162,也是大于0.05,得的结论与上面一致,即此次的成绩符合正态分布。
检验列联表数据
列联表是啥刚才已经解释过了,我们常见的就是2×2的列联表形式,还是吸烟与肺癌的例子:
代码实现如下:

所得的p值为0.004855,小于0.05,可以认为吸烟与患癌病有关。
检验列联表数据时,我们还常会用到Fisher检验,该检验是建立在超几何分布的基础上,当样本量比较小时(这里有个经验值,频数小于4),一般建议使用该检验方法。
Tips 1)超几何分布:是统计学上一种离散概率分布,描述的是,从有限个物件中抽出n个物件,
成功抽出指定类型的物件的次数(不归还)。比如,在产品质量的抽检中(抽出来的不放回去),
如果N件产品中有M件次品,抽检n件时所得次品数X,就服从超几何分布。 2)频数:也称“次数”,对整个数据按某种标准分组,然后统计出各个组内含个体的个数。
比如上面例子中,有一个频数为3(不吸烟患肺癌这一组),所以这里建议使用Fisher检验,其代码实现如下:
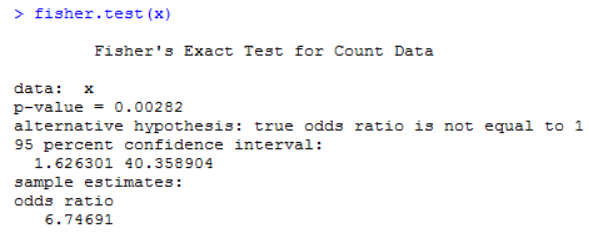
所得的p值为0.00282,小于0.05,所得结论与上面一致。且这里还给出了优势比(odds ratio)的值,该值大于1,其含义是吸烟越多,患肺癌的可能性就越大。
求得P值,下结论
其实在第2部分我们举例说明的时候就已经做了这一步。在这里,我们想着重说明一下的是:
a. 在我们下结论的时候,其实是用了一个大家广泛接受的原则:在一次试验中,小概率事件基本上不会发生。若它正好发生了,那么我们就有理由怀疑原假设的准确性;
b. 对于显著性水平a的大小(比如0.01, 0.05, 0.1之类),这个大小没有一个绝对的定值,只要你能说出一些合理的理由,你完全可以调整这个值的界限。若是对这些约定俗成的值感兴趣的,可以去看看那些老前辈的文章(比如《Statistical methods for research workers》);
c. 从前面描述,我们都可以看出,我们所下结论时都是根据p值而来,所以,在描述结论的时候一定不能下绝对的结论,比如“认为3组小鼠的平均存活天数一定(或肯定)有差异”,这种结论是不严谨的;
d. 我们所说的“具有显著性差异”,这个表现不出具体差异的大小,只能说明它们存在差异,所以,这也是我们在分析数据时,经常会看到大家不仅关注p值,还同时关注倍数(fold change)变化的原因,这种可视化表现形式尤其以火山图最为著名。
原文地址:http://www.cnblogs.com/rongdie/p/16787651.html