
1. 学习目标
- 讨论为什么归一化计数对于细胞之间的准确比较是必要的
- 解释如何通过主成分分析 (
PCA) 评估细胞之间的相似性
在获得高质量单细胞后,scRNA-seq分析工作流程的下一步是执行聚类。聚类的目标是将不同的细胞类型分成独特的细胞簇。为了进行聚类,确定了细胞间表达差异最大的基因。然后,使用这些基因来确定哪些相关基因组是造成细胞间表达差异最大的原因。
2. 计数归一化
第一个是计数归一化,这对于准确比较细胞(或样本)之间的基因表达至关重要。除了许多其他因素之外,每个基因的映射读数计数与RNA的表达成正比。归一化是缩放原始计数值的过程。以这种方式,细胞之间或细胞内的表达水平更具可比性。
归一化过程中经常考虑的主要因素有:
- 测序深度
考虑测序深度对于比较细胞之间的基因表达是必要的。在下面的示例中,每个基因在细胞 2 中的表达似乎都增加了一倍,但这是细胞 2 具有两倍测序深度的结果。
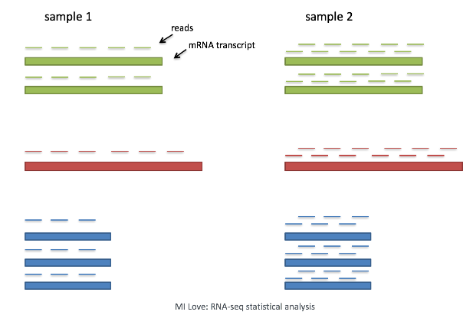
scRNA-seq 中的每个细胞都有不同数量的与其相关的读取。因此,为了准确比较细胞之间的表达,有必要对测序深度进行归一化。
- 基因长度
考虑基因长度对于比较同一细胞内不同基因之间的表达是必要的。映射到较长基因的读数的数量似乎与表达更高的较短基因具有相同的计数。
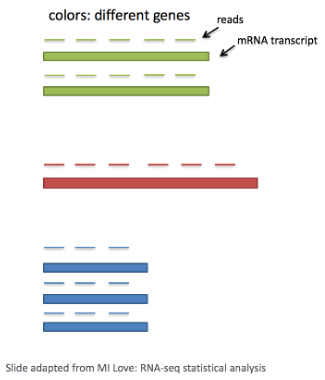
在scRNA-seq分析中,将比较细胞内不同基因的表达以对细胞进行聚类。如果使用基于 3′ 或 5′ 液滴的方法,基因的长度不会影响分析,因为仅对转录本的 5′ 或 3′ 端进行测序。但是,如果使用全长测序,则应考虑转录本长度。
3. PCA
主成分分析 (PCA) 是一种用于强调变化和相似性的技术,并在数据集中显示出强烈的模式;它是用于“降维”的方法之一。本课中简要介绍 PCA,强烈建议您浏览StatQuest 的视频以获得更全面的解释。
- 一个简单的例子
假设您已经量化了两个样本(或细胞)中四个基因的表达,您可以绘制这些基因的表达值,其中一个样本代表 x 轴,另一个样本代表 y 轴,如下所示:
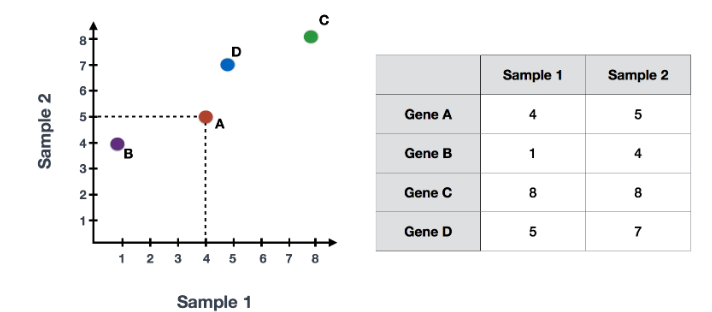
您可以在表示最大变化的方向上通过数据绘制一条线,在本例中位于对角线上。数据集中的最大变异发生在构成这条线的两个端点的基因之间。
基因在线上和线下有所不同。可以在数据中绘制另一条线,表示数据中第二大的变化量,因为该图是二维的(2 个轴)。

每行末端附近的基因是变异最大的基因;从数学上讲,这些基因对线的方向影响最大。
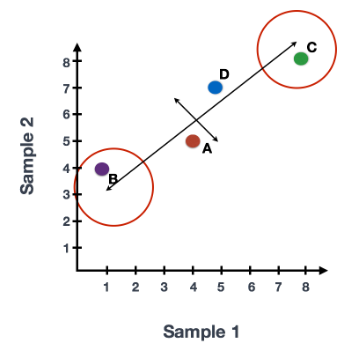
例如,Gene C 值的微小变化会极大地改变较长线的方向,而 Gene A 或 Gene D 的微小变化对其影响不大。
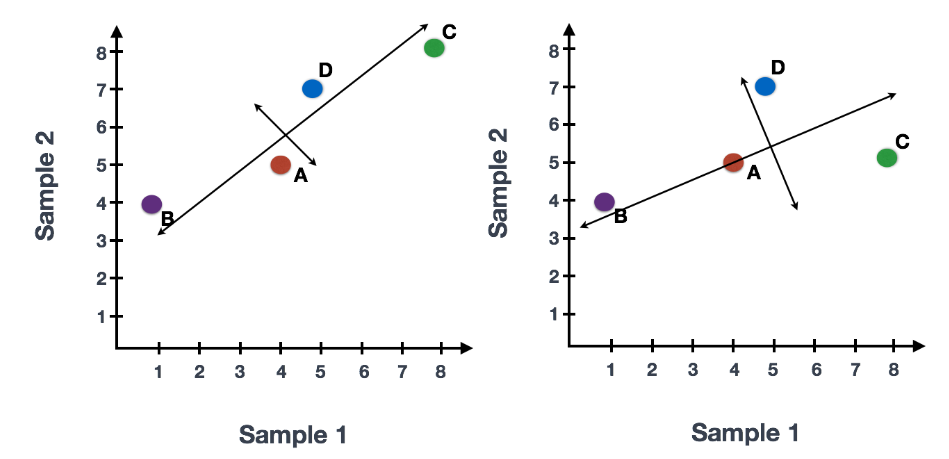
还可以旋转整个图,并查看代表变化的线条从左到右和上下。看到数据中的大部分变化是从左到右(较长的线),数据中第二大的变化是上下(较短的线)。您现在可以将这些线视为表示变化的轴。这些轴本质上是“主成分”,PC1 代表数据中最大的变化,PC2 代表数据中第二大的变化。
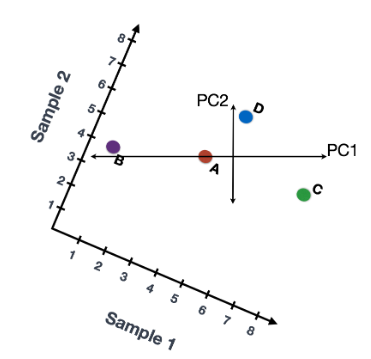
现在,如果有三个样本/细胞,那么将有一个额外的方向,可以在其中进行变化。因此,如果有 N 个样本/细胞,将有 N 个变化方向或 N 个主成分(PCs)!计算完这些 PC 后,处理数据集中变化最大的 PC 被指定为 PC1,下一个被指定为 PC2,以此类推。
一旦确定了数据集的 PC,必须弄清楚每个样本/单元如何重新适应该上下文,能够以直观的方式可视化相似性/不相似性。这里的问题是“基于 sample_X 中的基因表达,给定 PC 的 sample_X 得分是多少?”。这是降低维度的实际步骤,因为最终 PCA 图上绘制每个样本/单元格的 PC 分数。
为所有样本-PC 对计算 PC 分数,如下面的步骤和示意图中所述:
(1) 首先,根据每个基因对 PC 的影响程度,为每个基因分配一个“影响”分数。对给定 PC 没有任何影响的基因得分接近于零,而影响更大的基因得分更高。 PC 线末端的基因将产生更大的影响,因此它们将获得更大的分数,但符号相反。
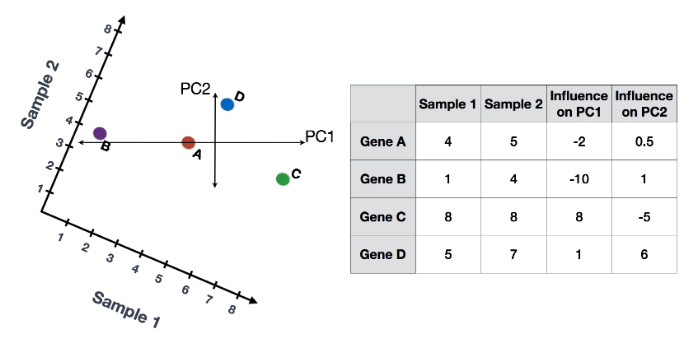
(2) 确定影响后,使用以下等式计算每个样本的分数:
Sample1 PC1 score = (read count * influence) + ... for all genes
对于 2 个样本示例,以下是计算分数的方式:
## Sample1
PC1 score = (4 * -2) + (1 * -10) + (8 * 8) + (5 * 1) = 51
PC2 score = (4 * 0.5) + (1 * 1) + (8 * -5) + (5 * 6) = -7
## Sample2
PC1 score = (5 * -2) + (4 * -10) + (8 * 8) + (7 * 1) = 21
PC2 score = (5 * 0.5) + (4 * 1) + (8 * -5) + (7 * 6) = 8.5
这是前两个步骤的示意图:

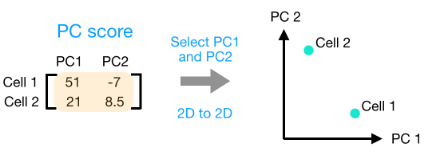
(3) 一旦为所有 PC 计算了这些分数,就可以将它们绘制在一个简单的散点图上。下面是此处示例的图,从 2D 矩阵到 2D 图:
scRNA-seq例子
假设您正在处理一个包含 12,000 个细胞的单细胞 RNA-seq 数据集,并且您已经量化了 20,000 个基因的表达。
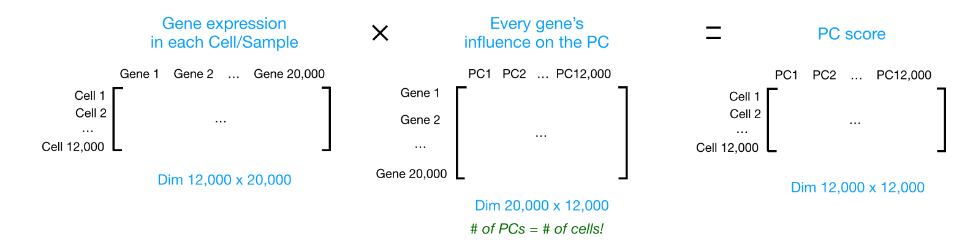
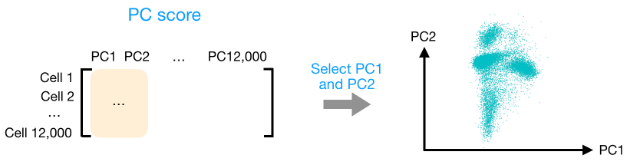
计算出 PC 分数后,您将看到一个 12,000 x 12,000 的矩阵,它表示有关所有细胞中相对基因表达的信息。您可以选择 PC1 和 PC2 列并以 2D 方式绘制。
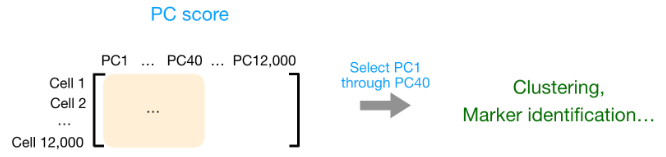
您还可以将前 40 台 PC 的 PC 分数用于聚类、标记识别等下游分析,因为这些代表了数据中的大部分变化。
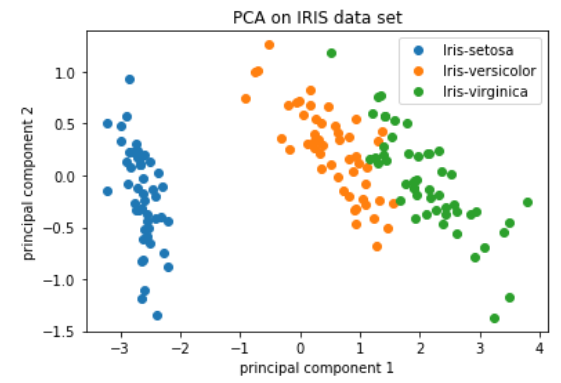
注意:对于具有大量样本或单元格的数据集,通常仅绘制每个样本/单元格的 PC1 和 PC2 分数,或用于可视化。由于这些 PC 解释了数据集中的最大变化,因此期望彼此更相似的样本/细胞将与 PC1 和 PC2 聚集在一起。请参阅下面的真实示例:
本文由mdnice多平台发布
原文地址:http://www.cnblogs.com/swindler/p/16867874.html